
Neurofibromatosis type 1
- Radiopaedia Australia Pty Ltd and Radiopaedia Events Pty Ltd, Director, Founder and CEO (Radiopaedia) (ongoing)
- Biogen Australia Pty Ltd, Investigator-Initiated Research Grant for CAD software in multiple sclerosis: finished Oct 2021 (past)
Updates to Article Attributes
Neurofibromatosis type 1 (NF1), also known as von Recklinghausen disease, is a multisystem neurocutaneous disorder, the most common phakomatosis, and a RASopathy. Additionally, it is also one of the most common inherited CNS disorders, autosomal dominant disorders, and inherited tumour syndromes.
Individual systemic manifestations are discussed individually:
- breast manifestations of NF1
- central nervous system manifestations of NF1
- cutaneous manifestations of NF1
- musculoskeletal manifestations of NF1
- pulmonary manifestations of NF1
- orbital manifestations of NF1
The remainder of this article pertains to a general discussion of neurofibromatosis type 1.
Epidemiology
Neurofibromatosis affects 1:2500-3000 individuals 3. In half of the cases, the disease is inherited as an autosomal dominant condition. In the other half, the disease is due to a de novo mutation 6. There is a variable expression but 100% penetrance by 5 years of age 6.
Clinical presentation
As is the case with many phakomatoses, NF1 results in a variety of abnormalities of variable severity. To make the clinical diagnosis two or more of the following are required 2:
- >6 cafe au lait spots evident during one year (prepubertal >0.5 cm, postpubertal >1.5 cm in size)
- two or more neurofibromas or one plexiform neurofibroma
- optic nerve glioma
- distinctive osseous lesion (such as sphenoid wing dysplasia or thinning of long bone cortex with or without pseudoarthrosis)
- two or more iris hamartomas (Lisch nodules)
- axillary or inguinal freckling
- a primary relative with NF1 with the above criteria
A mnemonic to help remember these features is CAFE SPOT.
In addition, ~45% (range 30-60%) of patients have learning disabilities, and approximately 1% have hypertension due to renal artery stenosis.
It is important to consider that NF1 has a much earlier age of onset than schwannomatosis and NF2, with approximately 50% of patients meeting the diagnostic criteria for NF1 by the age of 1 year and approximately 97% meeting the criteria by the age of 8 years 10.
Neoplasms
It should come as no surprise that a disease due to inactivation of a tumour suppressor gene (see below) is also associated with an increased incidence of numerous tumours 1-6:
- phaeochromocytoma
- malignant peripheral nerve sheath tumour (MPNST) (~10% of patients) 7
- Wilms tumour
- rhabdomyosarcoma
- renal angiomyolipoma
- glioma
- juvenile pilocytic astrocytoma (~20% of patients) 13
- optic nerve glioma
- diffuse brainstem glioma
- spinal astrocytoma and spinal pilocytic astrocytoma 9
- carcinoid tumour(s)
- leiomyoma(s)
- leiomyosarcoma
- ganglioglioma
- leukaemia
Pathology
The NF1 gene locus is on chromosome 17q11.2 and the gene product is neurofibromin, which acts as a tumour suppressor of the Ras/MAPK pathway; inactivation of the gene thus predisposes to tumour development 6,12,13. For this reason, the disorder is classified as a RASopathy 12.
The disease primarily is a hamartomatous disorder that involves the ectoderm and mesoderm. Usually, three types of neurofibromas occur in this disorder and are distinguished on the basis of their gross and microscopic appearances.
- localised neurofibroma (cutaneous neurofibroma): the most common type, is a focal lesion that typically is located in the dermis and subcutis
- diffuse neurofibroma (subcutaneous neurofibroma): localised in the subcutis, usually in the head and neck region.
- plexiform neurofibroma: considered pathognomonic if present; they may be seen in virtually any location but usually occur in the neck, pelvis, and extremities
Radiographic features
Breast
Central nervous system
- FASI (focal areas of signal intensity): occur in deep white matter and basal ganglia or corpus callosum 5, areas of T2/FLAIR hyperintensity with no contrast enhancement
- optic nerve glioma or optic pathway glioma (may manifest as enlarged optic foramen)
- progressive sphenoid wing dysplasia
- J-shaped sella
- lambdoid suture defects
- dural calcification at the vertex
- moya-moya phenomenon (rare)
- buphthalmos
Cutaneous
- cutaneous and subcutaneous neurofibromas: benign peripheral nerve sheath tumours
Skeletal
- kyphoscoliosis
- posterior vertebral scalloping
- hypoplastic posterior elements
- enlarged neural foramina
- ribbon rib deformity, rib notching, and dysplasia
- dural ectasia
- tibial pseudoarthrosis or, less commonly, ulnar pseudoarthrosis
- bony dysplasias: especially affecting the tibia
- severe bowing, gracile bones 11
- multiple non-ossifying fibromas
- limb hemihypertrophy
Thoracic
- mediastinal masses
- neurofibroma
- lateral thoracic meningocele: typically on the convex side of scoliosis (through widened neural foramina)
- extra-adrenal phaeochromocytoma
- lung parenchymal disease: ~20%
- diffuse interstitial fibrosis: lower zone
- bullae formation: upper zone
- secondary pulmonary arterial hypertension and cor pulmonale
Vascular
Treatment and prognosis
No single treatment exists, and a combination of supportive and surgical therapies are employed depending on the specific tumours and anomalies present.
Although prognosis is very variable, overall life expectancy is approximately half that of non-affected individuals. Tumours or cardiovascular complications are the most common causes of mortality 8.
History and etymology
The first name of this condition was von Recklinghausen disease because in 1882, Friedrich von Recklinghausen described cases of neurofibromatosis and recognised it as a nosological entity 14.
See also
-<p><strong>Neurofibromatosis type 1 (NF1)</strong>, also known as <strong>von Recklinghausen disease</strong>, is a multisystem neurocutaneous disorder, the most common <a href="/articles/phakomatoses">phakomatosis</a>, and a <a href="/articles/rasopathy-1">RASopathy</a>. Additionally, it is also one of the most common inherited CNS disorders, autosomal dominant disorders, and inherited tumour syndromes. </p><p>Individual systemic manifestations are discussed individually: </p><ul>-<li><a href="/articles/neurofibromatosis-type-1-breast-manifestations-1">breast manifestations of NF1</a></li>-<li><a href="/articles/neurofibromatosis-type-1-cns-manifestations-1">central nervous system manifestations of NF1</a></li>-<li><a href="/articles/neurofibromatosis-type-1-cutaneous-manifestations-1">cutaneous manifestations of NF1</a></li>-<li><a href="/articles/neurofibromatosis-type-1-musculoskeletal-manifestations-1">musculoskeletal manifestations of NF1</a></li>-<li><a href="/articles/pulmonary-manifestations-of-nf1">pulmonary manifestations of NF1</a></li>-<li><a href="/articles/neurofibromatosis-type-1-orbital-manifestations-2">orbital manifestations of NF1</a></li>-</ul><p>The remainder of this article pertains to a general discussion of neurofibromatosis type 1. </p><h4>Epidemiology</h4><p>Neurofibromatosis affects 1:2500-3000 individuals <sup>3</sup>. In half of the cases, the disease is inherited as an autosomal dominant condition. In the other half, the disease is due to a de novo mutation <sup>6</sup>. There is a variable expression but 100% penetrance by 5 years of age <sup>6</sup>.</p><h4>Clinical presentation</h4><p>As is the case with many phakomatoses, NF1 results in a variety of abnormalities of variable severity. To make the clinical diagnosis two or more of the following are required <sup>2</sup>:</p><ul>-<li>>6 <a href="/articles/cafe-au-lait-spots">cafe au lait spots</a> evident during one year (prepubertal >0.5 cm, postpubertal >1.5 cm in size)</li>-<li>two or more <a href="/articles/neurofibroma">neurofibromas</a> or one <a href="/articles/plexiform-neurofibroma">plexiform neurofibroma</a>-</li>-<li><a href="/articles/optic-pathway-glioma">optic nerve glioma </a></li>-<li>distinctive osseous lesion (such as <a href="/articles/sphenoid-wing-dysplasia">sphenoid wing dysplasia</a> or thinning of long bone cortex with or without <a href="/articles/congenital-pseudoarthrosis-of-the-tibia">pseudoarthrosis</a>)</li>-<li>two or more iris hamartomas (Lisch nodules) </li>-<li>axillary or inguinal freckling</li>-<li>a primary relative with NF1 with the above criteria</li>-</ul><p>A mnemonic to help remember these features is <a href="/articles/neurofibromatosis-type-1-mnemonic">CAFE SPOT</a>.</p><p>In addition, ~45% (range 30-60%) of patients have learning disabilities, and approximately 1% have hypertension due to <a href="/articles/renal-artery-stenosis">renal artery stenosis</a>.</p><p>It is important to consider that NF1 has a much earlier age of onset than schwannomatosis and <a href="/articles/neurofibromatosis-type-2-3">NF2</a>, with approximately 50% of patients meeting the diagnostic criteria for NF1 by the age of 1 year and approximately 97% meeting the criteria by the age of 8 years <sup>10</sup>. </p><h6>Neoplasms</h6><p>It should come as no surprise that a disease due to inactivation of a tumour suppressor gene (see below) is also associated with an increased incidence of numerous tumours <sup>1-6</sup>:</p><ul>-<li><a href="/articles/phaeochromocytoma-1">phaeochromocytoma</a></li>-<li>-<a href="/articles/malignant-peripheral-nerve-sheath-tumour">malignant peripheral nerve sheath tumour (MPNST)</a> (~10% of patients) <sup>7</sup>-</li>-<li><a href="/articles/wilms-tumour">Wilms tumour</a></li>-<li><a href="/articles/rhabdomyosarcoma">rhabdomyosarcoma</a></li>-<li><a href="/articles/renal-angiomyolipoma">renal angiomyolipoma</a></li>-<li>glioma<ul>-<li>-<a href="/articles/pilocytic-astrocytoma">juvenile pilocytic astrocytoma</a> (~20% of patients) <sup>13</sup>-</li>-<li><a href="/articles/optic-pathway-glioma">optic nerve glioma</a></li>-<li><a href="/articles/diffuse-brainstem-glioma-historical">diffuse brainstem glioma</a></li>-<li>-<a href="/articles/spinal-astrocytoma">spinal astrocytoma</a> and <a href="/articles/spinal-pilocytic-astrocytoma">spinal pilocytic astrocytoma</a> <sup>9</sup>-</li>-</ul>-</li>-<li><a href="/articles/carcinoid-tumour-2">carcinoid tumour(s)</a></li>-<li><a href="/articles/leiomyoma">leiomyoma(s)</a></li>-<li><a href="/articles/leiomyosarcoma">leiomyosarcoma</a></li>-<li><a href="/articles/ganglioglioma">ganglioglioma</a></li>-<li><a href="/articles/leukaemia">leukaemia</a></li>-</ul><h4>Pathology</h4><p>The NF1 gene locus is on chromosome 17q11.2 and the gene product is neurofibromin, which acts as a tumour suppressor of the <a href="/articles/mapk-pathway">Ras/MAPK pathway</a>; inactivation of the gene thus predisposes to tumour development <sup>6,12,13</sup>. For this reason, the disorder is classified as a <a href="/articles/rasopathy-1">RASopathy</a> <sup>12</sup>.</p><p>The disease primarily is a <a href="/articles/hamartoma">hamartomatous</a> disorder that involves the ectoderm and mesoderm. Usually, three types of neurofibromas occur in this disorder and are distinguished on the basis of their gross and microscopic appearances. </p><ul>-<li>-<a href="/articles/localised-neurofibroma">localised neurofibroma</a> (cutaneous neurofibroma): the most common type, is a focal lesion that typically is located in the dermis and subcutis</li>-<li>-<a href="/articles/diffuse-neurofibroma">diffuse neurofibroma</a> (subcutaneous neurofibroma): localised in the subcutis, usually in the head and neck region. </li>-<li>-<a href="/articles/plexiform-neurofibroma">plexiform neurofibroma</a>: considered pathognomonic if present; they may be seen in virtually any location but usually occur in the neck, pelvis, and extremities </li>-</ul><h4>Radiographic features</h4><h5>Breast</h5><ul><li><a href="/articles/neurofibromatosis-type-1-breast-manifestations-1">neurofibromatosis of the breast</a></li></ul><h5>Central nervous system</h5><ul>-<li>-<a href="/articles/fasi">FASI</a> (focal areas of signal intensity): occur in deep white matter and basal ganglia or corpus callosum <sup>5</sup>, areas of T2/FLAIR hyperintensity with no contrast enhancement</li>-<li>-<a href="/articles/optic-pathway-glioma">optic nerve glioma</a> or optic pathway glioma (may manifest as enlarged optic foramen)</li>-<li>progressive <a href="/articles/sphenoid-wing-dysplasia">sphenoid wing dysplasia </a>-</li>-<li><a href="/articles/j-shaped-sella-1">J-shaped sella</a></li>-<li>lambdoid suture defects</li>-<li>dural calcification at the vertex</li>-<li>-<a href="/articles/moyamoya-disease-1">moya-moya phenomenon</a> (rare)</li>-<li><a href="/articles/buphthalmos">buphthalmos</a></li>-</ul><h5>Cutaneous</h5><ul><li>cutaneous and subcutaneous <a href="/articles/localised-neurofibroma">neurofibromas</a>: benign peripheral nerve sheath tumours</li></ul><h5>Skeletal</h5><ul>-<li><a href="/articles/kyphoscoliosis">kyphoscoliosis</a></li>-<li>posterior <a href="/articles/vertebral-scalloping">vertebral scalloping</a>-</li>-<li>hypoplastic posterior elements</li>-<li><a href="/articles/enlarged-neural-foramina">enlarged neural foramina</a></li>-<li>-<a href="/articles/ribbon-rib-deformity">ribbon rib deformity</a>, <a href="/articles/rib-notching">rib notching</a>, and dysplasia </li>-<li><a href="/articles/dural-ectasia">dural ectasia</a></li>-<li>-<a href="/articles/congenital-pseudoarthrosis-of-the-tibia">tibial pseudoarthrosis</a> or, less commonly, ulnar pseudoarthrosis</li>-<li>bony dysplasias: especially affecting the tibia</li>-<li>severe bowing, <a href="/articles/gracile-bones">gracile bones</a> <sup>11</sup>-</li>-<li>multiple <a href="/articles/non-ossifying-fibroma-1">non-ossifying fibromas</a>-</li>-<li><a href="/articles/limb-hemi-hypertrophy">limb hemihypertrophy</a></li>-</ul><h5>Thoracic</h5><ul>-<li>mediastinal masses <ul>-<li><a href="/articles/neurofibroma">neurofibroma</a></li>-<li>-<a href="/articles/lateral-thoracic-meningocoeles">lateral thoracic meningocele</a>: typically on the convex side of scoliosis (through widened neural foramina)</li>-<li>extra-adrenal <a href="/articles/phaeochromocytoma-1">phaeochromocytoma</a>-</li>-</ul>-</li>-<li>lung parenchymal disease: ~20%<ul>-<li>-<a title="Pulmonary fibrosis" href="/articles/pulmonary-fibrosis">diffuse interstitial fibrosis</a>: lower zone</li>-<li>-<a href="/articles/pulmonary-bullae">bullae</a> formation: upper zone</li>-<li>secondary <a href="/articles/pulmonary-hypertension-1">pulmonary arterial hypertension</a> and <a href="/articles/cor-pulmonale-2">cor pulmonale</a>-</li>-</ul>-</li>-</ul><h5>Vascular</h5><ul>-<li>-<a href="/articles/aneurysm">aneurysms</a> / <a href="/cases/arteriovenous-malformation-cerebral-2">arteriovenous malformations</a>-</li>-<li><a href="/articles/renal-artery-stenosis">renal artery stenosis</a></li>-<li><a href="/cases/coarctation-of-aorta-3">coarctation of aorta</a></li>- +<p><strong>Neurofibromatosis type 1 (NF1)</strong>, also known as <strong>von Recklinghausen disease</strong>, is a multisystem neurocutaneous disorder, the most common <a href="/articles/phakomatoses">phakomatosis</a>, and a <a href="/articles/rasopathy-1">RASopathy</a>. Additionally, it is also one of the most common inherited CNS disorders, autosomal dominant disorders, and inherited tumour syndromes. </p><p>Individual systemic manifestations are discussed individually: </p><ul>
- +<li><a href="/articles/neurofibromatosis-type-1-breast-manifestations-1">breast manifestations of NF1</a></li>
- +<li><a href="/articles/neurofibromatosis-type-1-cns-manifestations-1">central nervous system manifestations of NF1</a></li>
- +<li><a href="/articles/neurofibromatosis-type-1-cutaneous-manifestations-1">cutaneous manifestations of NF1</a></li>
- +<li><a href="/articles/neurofibromatosis-type-1-musculoskeletal-manifestations-1">musculoskeletal manifestations of NF1</a></li>
- +<li><a href="/articles/pulmonary-manifestations-of-nf1">pulmonary manifestations of NF1</a></li>
- +<li><a href="/articles/neurofibromatosis-type-1-orbital-manifestations-2">orbital manifestations of NF1</a></li>
- +</ul><p>The remainder of this article pertains to a general discussion of neurofibromatosis type 1. </p><h4>Epidemiology</h4><p>Neurofibromatosis affects 1:2500-3000 individuals <sup>3</sup>. In half of the cases, the disease is inherited as an autosomal dominant condition. In the other half, the disease is due to a de novo mutation <sup>6</sup>. There is a variable expression but 100% penetrance by 5 years of age <sup>6</sup>.</p><h4>Clinical presentation</h4><p>As is the case with many phakomatoses, NF1 results in a variety of abnormalities of variable severity. To make the clinical diagnosis two or more of the following are required <sup>2</sup>:</p><ul>
- +<li>>6 <a href="/articles/cafe-au-lait-spots">cafe au lait spots</a> evident during one year (prepubertal >0.5 cm, postpubertal >1.5 cm in size)</li>
- +<li>two or more <a href="/articles/neurofibroma">neurofibromas</a> or one <a href="/articles/plexiform-neurofibroma">plexiform neurofibroma</a>
- +</li>
- +<li><a href="/articles/optic-pathway-glioma">optic nerve glioma </a></li>
- +<li>distinctive osseous lesion (such as <a href="/articles/sphenoid-wing-dysplasia">sphenoid wing dysplasia</a> or thinning of long bone cortex with or without <a href="/articles/congenital-pseudoarthrosis-of-the-tibia">pseudoarthrosis</a>)</li>
- +<li>two or more iris hamartomas (Lisch nodules) </li>
- +<li>axillary or inguinal freckling</li>
- +<li>a primary relative with NF1 with the above criteria</li>
- +</ul><p>A mnemonic to help remember these features is <a href="/articles/neurofibromatosis-type-1-mnemonic">CAFE SPOT</a>.</p><p>In addition, ~45% (range 30-60%) of patients have learning disabilities, and approximately 1% have hypertension due to <a href="/articles/renal-artery-stenosis">renal artery stenosis</a>.</p><p>It is important to consider that NF1 has a much earlier age of onset than schwannomatosis and <a href="/articles/neurofibromatosis-type-2-3">NF2</a>, with approximately 50% of patients meeting the diagnostic criteria for NF1 by the age of 1 year and approximately 97% meeting the criteria by the age of 8 years <sup>10</sup>. </p><h6>Neoplasms</h6><p>It should come as no surprise that a disease due to inactivation of a tumour suppressor gene (see below) is also associated with an increased incidence of numerous tumours <sup>1-6</sup>:</p><ul>
- +<li><a href="/articles/phaeochromocytoma-1">phaeochromocytoma</a></li>
- +<li>
- +<a href="/articles/malignant-peripheral-nerve-sheath-tumour">malignant peripheral nerve sheath tumour (MPNST)</a> (~10% of patients) <sup>7</sup>
- +</li>
- +<li><a href="/articles/wilms-tumour">Wilms tumour</a></li>
- +<li><a href="/articles/rhabdomyosarcoma">rhabdomyosarcoma</a></li>
- +<li><a href="/articles/renal-angiomyolipoma">renal angiomyolipoma</a></li>
- +<li>glioma<ul>
- +<li>
- +<a href="/articles/pilocytic-astrocytoma">juvenile pilocytic astrocytoma</a> (~20% of patients) <sup>13</sup>
- +</li>
- +<li><a href="/articles/optic-pathway-glioma">optic nerve glioma</a></li>
- +<li><a href="/articles/diffuse-brainstem-glioma-historical">diffuse brainstem glioma</a></li>
- +<li>
- +<a href="/articles/spinal-astrocytoma">spinal astrocytoma</a> and <a href="/articles/spinal-pilocytic-astrocytoma">spinal pilocytic astrocytoma</a> <sup>9</sup>
- +</li>
- +</ul>
- +</li>
- +<li><a href="/articles/carcinoid-tumour-2">carcinoid tumour(s)</a></li>
- +<li><a href="/articles/leiomyoma">leiomyoma(s)</a></li>
- +<li><a href="/articles/leiomyosarcoma">leiomyosarcoma</a></li>
- +<li><a href="/articles/ganglioglioma">ganglioglioma</a></li>
- +<li><a href="/articles/leukaemia">leukaemia</a></li>
- +</ul><h4>Pathology</h4><p>The NF1 gene locus is on chromosome 17q11.2 and the gene product is neurofibromin, which acts as a tumour suppressor of the <a href="/articles/mapk-pathway">Ras/MAPK pathway</a>; inactivation of the gene thus predisposes to tumour development <sup>6,12,13</sup>. For this reason, the disorder is classified as a <a href="/articles/rasopathy-1">RASopathy</a> <sup>12</sup>.</p><p>The disease primarily is a <a href="/articles/hamartoma">hamartomatous</a> disorder that involves the ectoderm and mesoderm. Usually, three types of neurofibromas occur in this disorder and are distinguished on the basis of their gross and microscopic appearances. </p><ul>
- +<li>
- +<a href="/articles/localised-neurofibroma">localised neurofibroma</a> (cutaneous neurofibroma): the most common type, is a focal lesion that typically is located in the dermis and subcutis</li>
- +<li>
- +<a href="/articles/diffuse-neurofibroma">diffuse neurofibroma</a> (subcutaneous neurofibroma): localised in the subcutis, usually in the head and neck region. </li>
- +<li>
- +<a href="/articles/plexiform-neurofibroma">plexiform neurofibroma</a>: considered pathognomonic if present; they may be seen in virtually any location but usually occur in the neck, pelvis, and extremities </li>
- +</ul><h4>Radiographic features</h4><h5>Breast</h5><ul><li><a href="/articles/neurofibromatosis-type-1-breast-manifestations-1">neurofibromatosis of the breast</a></li></ul><h5>Central nervous system</h5><ul>
- +<li>
- +<a href="/articles/fasi">FASI</a> (focal areas of signal intensity): occur in deep white matter and basal ganglia or corpus callosum <sup>5</sup>, areas of T2/FLAIR hyperintensity with no contrast enhancement</li>
- +<li>
- +<a href="/articles/optic-pathway-glioma">optic nerve glioma</a> or optic pathway glioma (may manifest as enlarged optic foramen)</li>
- +<li>progressive <a href="/articles/sphenoid-wing-dysplasia">sphenoid wing dysplasia </a>
- +</li>
- +<li><a href="/articles/j-shaped-sella-1">J-shaped sella</a></li>
- +<li>lambdoid suture defects</li>
- +<li>dural calcification at the vertex</li>
- +<li>
- +<a href="/articles/moyamoya-disease-1">moya-moya phenomenon</a> (rare)</li>
- +<li><a href="/articles/buphthalmos">buphthalmos</a></li>
- +</ul><h5>Cutaneous</h5><ul><li>cutaneous and subcutaneous <a href="/articles/localised-neurofibroma">neurofibromas</a>: benign peripheral nerve sheath tumours</li></ul><h5>Skeletal</h5><ul>
- +<li><a href="/articles/kyphoscoliosis">kyphoscoliosis</a></li>
- +<li>posterior <a href="/articles/vertebral-scalloping">vertebral scalloping</a>
- +</li>
- +<li>hypoplastic posterior elements</li>
- +<li><a href="/articles/enlarged-neural-foramina">enlarged neural foramina</a></li>
- +<li>
- +<a href="/articles/ribbon-rib-deformity">ribbon rib deformity</a>, <a href="/articles/rib-notching">rib notching</a>, and dysplasia </li>
- +<li><a href="/articles/dural-ectasia">dural ectasia</a></li>
- +<li>
- +<a href="/articles/congenital-pseudoarthrosis-of-the-tibia">tibial pseudoarthrosis</a> or, less commonly, ulnar pseudoarthrosis</li>
- +<li>bony dysplasias: especially affecting the tibia</li>
- +<li>severe bowing, <a href="/articles/gracile-bones">gracile bones</a> <sup>11</sup>
- +</li>
- +<li>multiple <a href="/articles/non-ossifying-fibroma-1">non-ossifying fibromas</a>
- +</li>
- +<li><a href="/articles/limb-hemi-hypertrophy">limb hemihypertrophy</a></li>
- +</ul><h5>Thoracic</h5><ul>
- +<li>mediastinal masses <ul>
- +<li><a href="/articles/neurofibroma">neurofibroma</a></li>
- +<li>
- +<a href="/articles/lateral-thoracic-meningocoeles">lateral thoracic meningocele</a>: typically on the convex side of scoliosis (through widened neural foramina)</li>
- +<li>extra-adrenal <a href="/articles/phaeochromocytoma-1">phaeochromocytoma</a>
- +</li>
- +</ul>
- +</li>
- +<li>lung parenchymal disease: ~20%<ul>
- +<li>
- +<a title="Pulmonary fibrosis" href="/articles/pulmonary-fibrosis">diffuse interstitial fibrosis</a>: lower zone</li>
- +<li>
- +<a href="/articles/pulmonary-bullae">bullae</a> formation: upper zone</li>
- +<li>secondary <a href="/articles/pulmonary-hypertension-1">pulmonary arterial hypertension</a> and <a href="/articles/cor-pulmonale-2">cor pulmonale</a>
- +</li>
- +</ul>
- +</li>
- +</ul><h5>Vascular</h5><ul>
- +<li>
- +<a href="/articles/aneurysm">aneurysms</a> / <a href="/cases/arteriovenous-malformation-cerebral-2">arteriovenous malformations</a>
- +</li>
- +<li><a href="/articles/renal-artery-stenosis">renal artery stenosis</a></li>
- +<li><a href="/cases/coarctation-of-aorta-3">coarctation of aorta</a></li>
Image 2 Pathology ( create )

Image 3 MRI (T2) ( update )

Image 4 MRI (T2) ( update )

Image 5 MRI (T1 C+ fat sat) ( update )

Image 6 CT ( update )

Image 7 MRI (T1 C+) ( update )

Image 8 MRI (T2) ( update )

Image 9 CT (non-contrast) ( update )

Image 10 Annotated image (Frontal) ( update )

Image 11 Ultrasound ( update )

Image 12 Annotated image ( update )

Image 13 X-ray (Coned frontal) ( update )
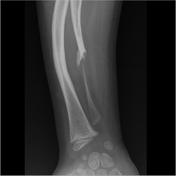
Image 14 MRI (T2 fat sat) ( update )

Image 15 MRI (T1 C+ fat sat) ( update )
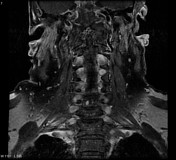
Image 16 Annotated image ( update )

Image 17 MRI (T1 C+ fat sat) ( update )
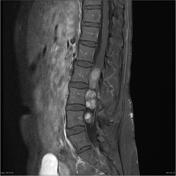
Image 18 MRI (T1) ( update )
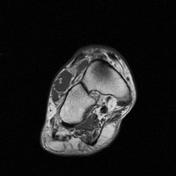
Image 19 MRI (T2 fat sat) ( update )

Image 20 CT (lung window) ( update )

Image 21 CT (non-contrast) ( update )
Image 22 CT (C+ portal venous phase) ( update )
Image 23 X-ray (Lateral) ( update )






 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.