
Thalassemia
Updates to Article Attributes
Thalassaemia is anautosomal recessive microcytic anaemia that originated in the Mediterranean region. The genetic defect causes a reduction in the rate of globin chain synthesis which causes the formation of abnormal haemoglobin molecules. The resultant anaemia is the characteristic presenting symptom of the thalassemias.
Thalassemia is a quantitative problem of globin synthesis, whereas sickle-cell disease (a haemoglobinopathy) is a qualitative problem of synthesis of an incorrectly functioning globin.
Pathophysiology
Normal adult haemoglobin is composed of Hb A (98%) and Hb A2 (2%) which contain two. Hb A contains two α globin chains /two/ two β globin chains, and HbA2 contains twoα globin chains/ twoδglobin chains, respectively. They are arranged arranged into a heterotetramer. Thalassaemia patients produce a deficiency of either α or β globin, unlike sickle-cell disease, which produces a specific mutant form of β globin.
The thalassemias are classified according to which chain of the haemoglobin molecule is affected. In α thalassemias, production of the α globin chain is reduced, while in β thalassemia production of the β globin chain is reduced.
The β globin chains are encoded by a single gene on chromosome 11; α globin chains are encoded by two closely linked genes on chromosome 16. Thus, in a normal person with two copies of each chromosome, there are two loci encoding the β chain, and four loci encoding the α chain. Deletion of one of the α loci has a high prevalence in people of African or Asian descent, making them more likely to develop α thalassemias. β Thalassemiasthalassemias are common in Africans, but also in Greeks and Italians.
The thalassemia trait may confer a degree of protection against malaria, which confers a selective survival advantage on carriers.
Radiographic features
Skeletal
Marrow proliferation consists of expansion of the medulla, thinning of cortical bone, and resorption of cancellous bone resulting in a a generalized loss of bone density.
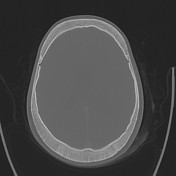
- skull: classic "hair-on-end" appearance
- facial bones: rodent facies
- ribs: "rib-within-a-rib" appearance, noted particularly in the middle and anterior portions of the ribs
- extramedullary hematopoiesis
- premature fusion of the epiphyses
Gastrointestinal: hepatobilliary
-<p><strong>Thalassaemia</strong> is an <a href="/articles/autosomal-recessive">autosomal recessive</a> <a href="/articles/microcytic-anaemia">microcytic anaemia</a> that originated in the Mediterranean region. The genetic defect causes a reduction in the rate of <a href="/articles/globin-chain">globin chain</a> synthesis which causes the formation of abnormal <a href="/articles/haemoglobin">haemoglobin </a>molecules. The resultant anaemia is the characteristic presenting symptom of the thalassemias.</p><p>Thalassemia is a quantitative problem of globin synthesis, whereas <a href="/articles/sickle-cell-disease">sickle-cell disease</a> (a <a href="/articles/haemoglobinopathy">haemoglobinopathy</a>) is a qualitative problem of synthesis of an incorrectly functioning globin.</p><h4>Pathophysiology</h4><p>Normal adult haemoglobin is composed of Hb A (98%) and Hb A2 (2%) which contain two <a href="/articles/a-globin-chains">α globin chains</a> /two <a href="/articles/b-globin-chains">β globin chains</a> and two <a href="/articles/a-globin-chains">α globin chains</a>/ two <a href="/articles/globin-chains">δ</a><a href="/articles/a-globin-chains"> </a><a href="/articles/globin-chains">globin chains</a>, respectively. They are arranged into a heterotetramer. Thalassaemia patients produce a deficiency of either α or β globin, unlike sickle-cell disease, which produces a specific mutant form of β globin.</p><p>The thalassemias are classified according to which chain of the haemoglobin molecule is affected. In α thalassemias, production of the α globin chain is reduced, while in β thalassemia production of the β globin chain is reduced.</p><p>The β globin chains are encoded by a single gene on chromosome 11; α globin chains are encoded by two closely linked genes on chromosome 16. Thus, in a normal person with two copies of each chromosome, there are two loci encoding the β chain, and four loci encoding the α chain. Deletion of one of the α loci has a high prevalence in people of African or Asian descent, making them more likely to develop α thalassemias. β Thalassemias are common in Africans, but also in Greeks and Italians.</p><p>The <a href="/articles/thalassemia-trait">thalassemia trait</a> may confer a degree of protection against malaria, which confers a selective survival advantage on carriers.</p><h4>Radiographic features</h4><h5>Skeletal</h5><p>Marrow proliferation consists of expansion of the medulla, thinning of <a href="/articles/cortical-bone">cortical bone</a>, and resorption of <a href="/articles/cancellous-bone">cancellous bone</a> resulting in a generalized loss of bone density.</p><ul>- +<p><strong>Thalassaemia</strong> is an <a href="/articles/autosomal-recessive">autosomal recessive</a> <a href="/articles/microcytic-anaemia">microcytic anaemia</a> that originated in the Mediterranean region. The genetic defect causes a reduction in the rate of <a href="/articles/globin-chain">globin chain</a> synthesis which causes the formation of abnormal <a href="/articles/haemoglobin">haemoglobin </a>molecules. The resultant anaemia is the characteristic presenting symptom of the thalassemias.</p><p>Thalassemia is a quantitative problem of globin synthesis, whereas <a href="/articles/sickle-cell-disease">sickle-cell disease</a> (a <a href="/articles/haemoglobinopathy">haemoglobinopathy</a>) is a qualitative problem of synthesis of an incorrectly functioning globin.</p><h4>Pathophysiology</h4><p>Normal adult haemoglobin is composed of Hb A (98%) and Hb A2 (2%). Hb A contains two <a href="/articles/a-globin-chains">α globin chains</a> / two <a href="/articles/b-globin-chains">β globin chains</a>, and HbA2 contains two <a href="/articles/a-globin-chains">α globin chains</a> / two <a href="/articles/globin-chains">δ</a><a href="/articles/a-globin-chains"> </a><a href="/articles/globin-chains">globin chains</a>. They are arranged into a heterotetramer. Thalassaemia patients produce a deficiency of either α or β globin, unlike sickle-cell disease, which produces a specific mutant form of β globin.</p><p>The thalassemias are classified according to which chain of the haemoglobin molecule is affected. In α thalassemias, production of the α globin chain is reduced, while in β thalassemia production of the β globin chain is reduced.</p><p>The β globin chains are encoded by a single gene on chromosome 11; α globin chains are encoded by two closely linked genes on chromosome 16. Thus, in a normal person with two copies of each chromosome, there are two loci encoding the β chain, and four loci encoding the α chain. Deletion of one of the α loci has a high prevalence in people of African or Asian descent, making them more likely to develop α thalassemias. β thalassemias are common in Africans, but also in Greeks and Italians.</p><p>The <a href="/articles/thalassemia-trait">thalassemia trait</a> may confer a degree of protection against malaria, which confers a selective survival advantage on carriers.</p><h4>Radiographic features</h4><h5>Skeletal</h5><p>Marrow proliferation consists of expansion of the medulla, thinning of <a href="/articles/cortical-bone">cortical bone</a>, and resorption of <a href="/articles/cancellous-bone">cancellous bone</a> resulting in a generalized loss of bone density.</p><ul>
Image ( destroy )
Image 4 Annotated image ( update )

Image 5 MRI (PD fat sat) ( update )

Image 6 X-ray (Frontal) ( update )

Image 7 X-ray (PA) ( update )

Image 8 CT (bone window) ( update )





 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.